Welcome to Nailpolish's documentation!
Nailpolish is a high-performance Rust tool designed to improve the accuracy of sequencing data by error correcting PCR duplicates.
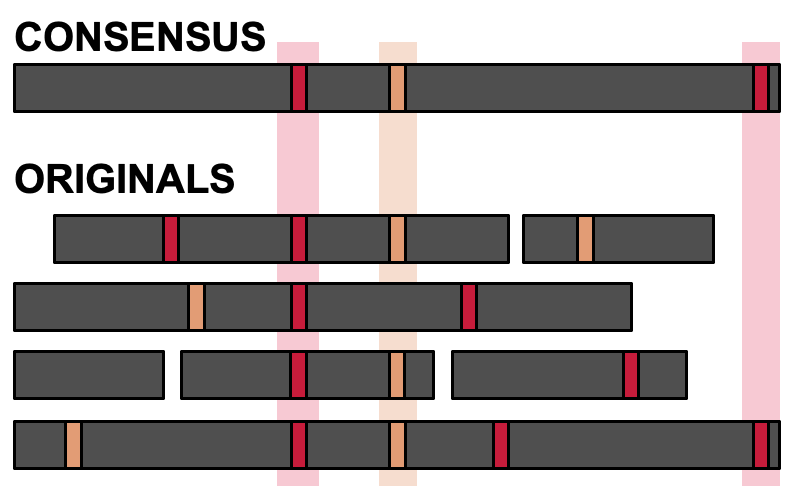
Nailpolish identifies PCR duplicates in barcoded data (reads containing identical barcodes and UMIs, forming "duplicate groups") and applies the partial order alignment consensus algorithm to replace multiple duplicate reads with a single consensus error-corrected read. This process corrects sequencing errors which naturally occur in the reads, improving the overall quality and reliability of sequencing data.
Nailpolish operates in a reference-free manner, first identifying duplicate groups and then clustering within each duplicate group. This process ensures that only true duplicates are included in consensus calling. That is, unrelated reads that share barcodes and UMIs (due to read or demultiplexing errors) are not consensus called together, and are instead separated into separate clusters.
See the Quick Start guide to begin using Nailpolish with your data.
Quick Start
Warning
This guide was written for Nailpolish v0.1.0 and so is considered depreciated. Commands listed here likely will not work.
This quick start guide will walk you through installing Nailpolish and running it on a small demo dataset. The demo dataset is a small subset of the scmixology2 Chromium 10x droplet-based dataset, sequenced using Nanopore technology, released by Tian et al. (2021).
Our Flexiplex tool is used to demultiplex the dataset.
Install
For more information, see Install.
For x64 Linux, run:
curl --proto '=https' --tlsv1.2 -LsSf "https://github.com/DavidsonGroup/nailpolish/releases/download/nightly_develop/nailpolish" -o nailpolish
chmod +x nailpolish
Get test files
Download the scmixology2 subset reads using:
wget https://github.com/DavidsonGroup/nailpolish/releases/download/sample-fastq-for-quickstart/scmixology2_sample.fastq
Indexation
For more information, see nailpolish index.
By default, nailpolish expects the barcode and UMI to be in the @BC_UMI format at the start of the header.
Alternative barcode and UMI formats can be provided through either a preset (one of bc-umi, umi-tools, illumina)
or a custom barcode regex.
# write the index file to `index.tsv`
nailpolish index --index index.tsv scmixology2_sample.fastq
Summary of duplicate count
For more information, see nailpolish summary.
A .html file can be generated to summarise some key statistics about the input reads.
The output file is written to summary.html by default.
nailpolish summary index.tsv

Consensus call duplicates
For more information, see nailpolish call.
By consensus calling duplicates, only one read is returned per UMI group. For singleton reads, there is no change (apart from including UMI group information in the header).
nailpolish call \
--index index.tsv \
--input scmixology2_sample.fastq \
--output scmixology2_sample_consensus_called.fastq \
--threads 4
There are alternative parameters which can be passed to configure the output. See the nailpolish call documentation for more.
nailpolish index
This command is used to create an index file from a demultiplexed .fastq.
An index is required to run the other nailpolish commands.
The index command supports reads in multiple formats.
Usage
An index file will be created at <file>.fastq.nailpolish.idx.
$ nailpolish index --help
Create an index file from a demultiplexed .fastq
Usage: nailpolish index [OPTIONS] <INPUT> [PRESET]
Arguments:
<INPUT>
the input .fastq file
[PRESET]
[default: bc-umi]
Possible values:
- bc-umi: @BARCODE_UMI format as produced by Flexiplex for 10x3 chemistry
- umi-tools: `_<UMI>` format as produced by `umi-tools extract`
- illumina: bcl2fastq format, which has `:<UMI>` at the end of the read ID
Options:
--overwrite
overwrite an existing index file, if it exists
--clusters <CLUSTERS>
whether to use a file containing pre-clustered reads, with every line in one of two formats:
1. READ_ID;BARCODE
2. READ_ID;BARCODE;UMI
--barcode-regex <BARCODE_REGEX>
barcode regex format type, for custom header styles. this will override the preset given.
for example, for the `bc-umi` preset:
^([ATCG]{16})_([ATCG]{12})
--skip-unmatched
skip, instead of error, on reads which are not accounted for:
- if a cluster file is passed, any reads which are not in any cluster
- if a barcode regex or preset is used (default), any reads which do not match the regex
--len <LEN>
filter lengths to a value within the given float interval [a,b].
a is the minimum, and b is the maximum (both inclusive).
alternatively, a can be `-inf` and b can be `inf.
an unbounded interval (i.e. no length filter) is given by `0,inf`.
[default: 0,15000]
--qual <QUAL>
filter average read quality to a value within the given float interval [a,b].
see the docs for `--len` for documentation on how to use the interval.
[default: 0,inf]
-h, --help
Print help (see a summary with '-h')
Reading the index
Presets
Three presets are bundled with nailpolish for common barcode formats. These are useful when the header of each read contains information about the barcode.
bc-umi: read headers look like this:ATCGATCGATCG_ATCGATCGATCGATCGin theBC_UMIformat. This is the default barcoding format produced by the Flexiplex demultiplexer (Cheng et al. 2024).umi-tools: read headers look like this:HISEQ:87:00000000T_ATCGATCGATCGwhereATCGATCGATCGis the UMI sequence. This is the default UMI header format expected from the umi-tools (Smith et al. 2017) collection of UMI management tools.illumina: read headers look like this:SIM:1:FCX:1:2106:15337:1063:ATCGATCGATCG 1:N:0:ATCACGwhereATCGATCGATCGis the UMI sequence. This is the default UMI header format produced by tools such asbcl2fastq.
Barcode regex
For reads where barcodes and UMIs are contained in the header, in an esoteric format, a custom regular expression
can be provided through the --barcode-regex <BARCODE_REGEX> parameter. As examples, here are the regular expressions
for the presets above:
bc-umi:--barcode-regex "^([ATCG]{16})_([ATCG]{12})"umi-tools:--barcode-regex "_([ATCG]+)$"illumina:--barcode-regex ":([ATCG]+)$"
Regular expressions are parsed by the excellent regex library for Rust.
This library is performant and has guarantees on worst-case time complexity;
however, the scope of supported regular expression features is more limited.
For complex queries, it is recommended that you consult the crate documentation
and test your regular expression using regex101, ensuring that you set the 'Flavor' to 'Rust'.
nailpolish expects that every read in the input .fastq must be able to be matched to the provided
regular expression.
Cluster file
nailpolish can alternatively extract UMIs from a separately provided delimiter-separated file, if this information is
not in the read headers.
The file must be semicolon-delimited (;). Rows must be in the format READ_ID;BARCODE or READ_ID;BARCODE;UMI.
Note that no header line should be present in the file.
By default, nailpolish expects that every read in the input .fastq must have a corresponding entry in the
cluster file.
In the event where this is not the case, nailpolish will error. To ignore this error and silently skip over any
unmatched reads, the --skip-unmatched flag should be passed.
Filtering
nailpolish has filter settings which will exclude a read from being consensus called or considered part of a group. The read will be in the final consensus called output. This exists because sometimes sequencing errors can be excessively long, which have an outsized impact overall consensus calling time.
The default filtering settings are very conservative, only filtering reads with length >15000bp.
Two types of metadata can be filtered against: sequence length (using --len) and sequence quality (using --qual).
Filters should be intervals; that is, a string [a, b] where a and b represent the inclusive upper and lower
bound respectively.
nailpolish summary
Quickly review the quality and duplicate rate of the dataset. The reads must first have been indexed.
Usage
$ nailpolish summary --help
Generate a summary of duplicate statistics from an index file
Usage: nailpolish summary [OPTIONS] <INPUT>
Arguments:
<INPUT> Input .fastq file
Options:
-o, --output <OUTPUT> Output .html file. By default, will write to <file>.summary.html
-h, --help Print help
Output
See an example summary output file.
nailpolish consensus
Consensus call duplicated reads. The reads must first have been indexed. By default, reads within each duplicate group will be clustered to eliminate false duplicates.
Usage
$ nailpolish call --help
Generate a consensus-called 'cleaned up' file
Usage: nailpolish consensus [OPTIONS] <INPUT>
Arguments:
<INPUT> the input .fastq
Options:
-o, --output <OUTPUT> the output .fastq, or empty for stdout
-t, --threads <THREADS> the number of threads to use [default: 4]
--report-original-reads for each duplicate group of reads, report the original reads along with the consensus
--report-original-header if the original read headers are valuable, this will create a orig_header field
in the consensus called result with the entire original read header
--extra-stats add debugging information to the read header [intended for internal development]
warning: since timings are reported, the output will not be identical across runs
--no-clustering disable the clustering algorithm this will prevent nailpolish from detecting and separating false duplicates
-h, --help Print help
Output format
A .fastq file will be produced. By default, each read will look like this:
@GATAGCTAGCAACAAT_ATTTTACCGACC|id=12047|type=consensus|cluster=1|reads_called=2
# barcode─┘ UMI─┘ group id─┘ type─┘ │ │
# ID of cluster within group─┘ │
# this cluster has two reads in it─┘
# if duplicate group 12047 has 2 clusters, both are reported...
@GATAGCTAGCAACAAT_ATTTTACCGACC|id=12047|type=consensus|cluster=2|reads_called=3
# same group as above...─┘ │ │
# ...but different cluster─┘ │
# this cluster has three reads in it─┘
@GCAGTTAAGGATATAC_ACAGTTTCTTTG|id=2829|type=single|cluster=1|reads_called=1
# this group has only one read─┘ │ │
# in this case, these are always 1─┴──────────────┘
Flags can be passed to add other information to the output as well.
# using `--report-original-reads`, the original reads are produced as well...
@CTCAAGACATTGAGCT_ATTTTTTTTTTT|id=3566|type=original|read=3|cluster=2
# this is the original read─┘ │ │
# third read in the group─┘ │
# this read contributed to cluster 2─┘
# using `--report-original-header`, the original headers are outputted...
@GGAGGATTCTTCTAAC_TGTTCTTGAAGC|<...removed...>|orig_header=["GGAGGATTCTTCTAAC_TGTTCTTGAAGC#682b2274-473a-4a59-affe-30dbe4f1d070_+1of1","GGAGGATTCTTCTAAC_TGTTCTTGAAGC#36a1b7bd-5bab-45e1-a591-cb966f890f90_-1of1"]
# ... see the original header → → → → → ... these are the two reads that were called.
Options
--threads: set the number of threads that nailpolish should use--report-original-reads: report the original reads as well as the consensus read--report-original-header: report the original headers of the reads used to produce a consensus