Contents
- What does flexiplex do?
- Installing flexiplex
- Usage
- Examples of use
- Full long-read single-cell RNA-Seq tutorial
- Assigning long reads to 10x barcodes (when barcodes are known)
- Assigning long reads to 10x barcodes (when barcodes are unknown)
- Demultiplexing other read data by barcode
- NEW Demutiplexing multiple barcodes
- Assigning genotype to cells - long reads
- Assigning genotype to cells - short reads
- Simple search
- Extracting UMIs from PCR-cDNA ONT data
- Output
- Tips to speed up flexiplex
- Support or Contact
- Publication/Citation
What does flexiplex do?
Flexiplex is a light weight, flexible, error tolerant search and demultiplexing tool. Given a set of reads as either .fastq or .fasta it will demultiplex and/or identify target sequences, reporting matching reads and read-barcode assignment. It has been designed to demultiplex single cell long read RNA-Seq data, but can be used on any read data like an error tolerance “grep”. Flexiplex is built with edlib.
Flexiplex first uses edlib to search for a left and right flanking sequence (primer and polyT by default) within each read (with barcode and UMI sequence left as a wildcard). For the best match with an edit distance of “f” or less it will trim to the barcode + UMI sequence +/- 5 bp either side, and search for the barcode against a known list. The best matching barcode with an edit distance of “e” or less will be reported. Occassionally reads are chimeric, meaning two or more molecules get sequence togther in the same read. To identify these cases, and chop reads, flexiplex will repeat the search again with the previously found primer to polyT sequence masked out. This is repeated until no new barcodes are found in the read.
If the set of possible barcodes is unknown, flexiplex can be run in discovery mode (by leaving -k option off). In this mode, flexiplex will search for the primer and polyT sequence like usual, and take 16bp (by default) after the primer sequence as a barcode. The frequency that barcodes are found in the data are reported for follow up analysis. For example, by filtering with flexiplex-filter to obtain a final list of barcodes.
The primer, polyT, list of barcodes, UMI pattern and the order of these sequences can be adjusted through user settings. As can the maximum edit distances (see Usage).

Installing flexiplex
Precompiled binaries
Pre-compiled binaries are available in the Releases for Linux (x64) and Mac (Apple Silicon).
To run flexiplex-filter, it is easiest to use the uv package manager, which handles all dependencies in a temporary virtual environment:
uvx --from git+https://github.com/davidsongroup/flexiplex.git#subdirectory=scripts \
flexiplex-filter --help
For all the invocations of flexiplex-filter in this documentation, you can run it using uv as above.
Alternatively, you can install inside a virtual environment. flexiplex-filter has been tested on Python 3.9, but should work for relatively modern versions of Python.
# change into the scripts directory
cd <path_to_flexiplex_dir>/scripts
# create virtual environment
python -m venv .venv
# activate virtual environment (you must do this every time you run flexiplex-filter)
source .venv/bin/activate
# install
pip install .
# now you can run:
flexiplex-filter <parameters>
Compiling from source
# download from GitHub
wget https://github.com/DavidsonGroup/flexiplex/releases/download/v<x.y>/flexiplex-<x.y>.tar.gz
# untar and unzip
tar -xvf flexiplex-<x.y>.tar.gz
# change into source directory, compile, and install
cd flexiplex-<x.y>
make
Replacing
Alternatively, you can copy the binary to /usr/local/bin with make install.
Then install flexiplex-filter as before, using uv or:
# change into the scripts directory
cd <path_to_flexiplex_dir>/scripts
# create virtual environment
python -m venv .venv
# activate virtual environment (you must do this every time you run flexiplex-filter)
source .venv/bin/activate
# install
pip install .
# now you can run:
flexiplex-filter <parameters>
Conda
Alternatively, on Linux and non-Apple Silicon Macs, conda (or mamba) can be used to install flexiplex and flexiplex-filter together.
# optional, but recommended: install into a separate environment
conda create -c bioconda -c conda-forge --name flexiplex flexiplex
conda activate flexiplex
# alternatively, install directly into base
conda install -c bioconda -c conda-forge flexiplex
To see usage information, run
./flexiplex -h
Usage
FLEXIPLEX 1.02.6
usage: flexiplex [options] [reads_input]
reads_input: a .fastq or .fasta file. Will read from stdin if empty.
options:
-k known_list Either 1) a text file of expected barcodes in the first column,
one row per barcode, or 2) a comma separate string of barcodes.
Without this option, flexiplex will search and report possible barcodes.
The generated list can be used for known_list in subsequent runs.
-l true/false Replace read ID with barcodes+UMI (default: true)
-r true/false Remove search strings including flanking sequence and split read
if multiple barcodes found (default: true).
-s true/false Sort reads into separate files by barcode (default: false)
-c true/false Add a _C suffix to the read identifier of any chimeric reads
(default: false). For instance if,
@BC_UMI#READID_+1of2
is chimeric, it will become:
@BC_UMI#READID_+1of2_C
-n prefix Prefix for output filenames.
-e N Maximum edit distance to barcode (default 2).
-f N Maximum edit distance to primer+polyT (default 8).
-p N Number of threads (default: 1).
Specifying adaptor / barcode structure :
-x sequence Append flanking sequence to search for
-b sequence Append the barcode pattern to search for
-u sequence Append the UMI pattern to search for
Notes:
The order of these options matters
? - can be used as a wildcard
When no search pattern x,b,u option is provided, the following default pattern is used:
primer: CTACACGACGCTCTTCCGATCT
barcode: ????????????????
UMI: ????????????
polyT: TTTTTTTTT
which is the same as providing:
-x CTACACGACGCTCTTCCGATCT -b ???????????????? -u ???????????? -x TTTTTTTTT
Predefined search schemes:
-d 10x3v2 10x version 2 chemistry 3', equivalent to:
-x CTACACGACGCTCTTCCGATCT -b ???????????????? -u ?????????? -x TTTTTTTTT -f 8 -e 2
-d 10x3v3 10x version 3 chemistry 3', equivalent to:
-x CTACACGACGCTCTTCCGATCT -b ???????????????? -u ???????????? -x TTTTTTTTT -f 8 -e 2
-d 10x5v2 10x version 2 chemistry 5', equivalent to:
-x CTACACGACGCTCTTCCGATCT -b ???????????????? -u ?????????? -x TTTCTTATATGGG -f 8 -e 2
-d 10x_atac 10x ATAC-Seq, equivalent to:
-x ACCGAGATCTACAC -b ???????????????? -x CGCGTCTGTCGTCGGCAGCGTCAGATGTGTATAAGAGACAG -f 8 -e 2
-d grep Simple grep-like search (edit distance up to 2), equivalent to:
-f 2 -k ? -b '' -u '' -l false -r false
-h Print this usage information.
Have a different barcode scheme you would like Flexiplex to work with? Post a request at:
https://github.com/DavidsonGroup/flexiplex/issues
If you use Flexiplex in your research, please cite our paper:
O. Cheng et al., Flexiplex: a versatile demultiplexer and search tool for omics data, Bioinformatics, Volume 40, Issue 3, 2024
Examples of use
Full long-read single-cell RNA-Seq tutorial
Now available at https://davidsongroup.github.io/flexiplex/tutorial.html, which shows how flexiplex can be used in combination with other tools to go from a fastq file to cleaned and deduplicated UMI count matrix.
Assigning long reads to 10x barcodes (when barcodes are known)
There are several pre-sets which work for various 10x chemistry:
flexiplex -d <chemistry> -k barcode_list.txt reads.fastq > new_reads.fastq
10x 3’ version 3:
flexiplex -d 10x3v3 -k barcode_list.txt reads.fastq > new_reads.fastq
10x 3’ version 2 (10bp UMIs):
flexiplex -d 10x3v2 -k barcode_list.txt reads.fastq > new_reads.fastq
10x 5’ version 2:
flexiplex -d 10x5v2 -k barcode_list.txt reads.fastq > new_reads.fastq
Visium spatial (this has an identical barcode structure to 3’ version 3):
flexiplex -d 10x3v3 -k spot_barcode_list.txt reads.fastq > new_reads.fastq
If dealing with large gzipped files you can pipe reads into flexiplex to avoid unzipping. e.g.
gunzip -c read.fastq.gz | flexiplex -d 10x3v3 -k barcode_list.txt | gzip > new_reads.fastq.gz
Assigning long reads to 10x barcodes (when barcodes are unknown)
Flexiplex can be run in two passes: 1) to find the barcode sequences and 2) assign them to reads. To find barcodes, set the flanking edit distance to 0 (a perfect match) as these are less likely to have errors in the barcodes:
flexiplex -d <chemistry> -f 0 reads.fastq
e.g.
flexiplex -d 10x3v3 -f 0 reads.fastq
or for a zipped fastq:
gunzip -c read.fastq.gz | flexiplex -d 10x3v3 -f 0
Flexiplex will output a table which gives the frequency of how often each barcode is observed in the data. This table will need to be filtered for high quality barcodes which are free from errors. Do automate this, Flexiplex comes bundled with a standalone python script, flexiplex-filter. Flexiplex-filter can also filter against any list of possible barcodes, such as the possible 10x barcodes.
flexiplex-filter --whitelist 3M-february-2018.txt --no-inflection --outfile my_filtered_barcode_list.txt my_barcode_list.txtThis script also allows for the discovery and visualisation of points of inflection in a single cell knee plot. A brief ‘autopilot’ mode is provided which will determine an inflection point and filter out any cells with a lower count:
flexiplex-filter --whitelist 3M-february-2018.txt --outfile my_filtered_barcode_list.txt my_barcode_list.txtThe usage guide explains how to further use
filter-filterto visualise and fine-tune the inflection point result.
Then use this list to assign barcodes to reads:
flexiplex -d <chemistry> -k my_filtered_barcode_list.txt reads.fastq > new_reads.fastq
Demultiplexing barcodes from the beginning or end of reads
A common scenario is for no flanking sequence to be known, but the barcode to begin or end a fixed number of bases from the start or end of a read. In this instance you can add sequence to the beginnning/end to anchor the search. e.g. using “START” to anchor:
cat file.fastq | sed "/^[@+]/! s/^/START/g" | flexiplex -x "START" -f 0 -b "????????????????" -e 1 -k my_barcode_list.txt
Would search for 16bp barcodes from my_barcode_list.txt directly at the start of reads with an edit distance of 1.
Extracting UMIs from PCR-cDNA ONT data
Recent library prepration kits for ONT PCR amplified cDNA attach unique molecular identifiers (UMIs) to the 5’ end of transcripts. These can be used for deduplication in downstream data analysis. The UMI sequence for each read can be extracted using flexiplex:
flexiplex -x TTGGTGCTGATATTGCTTTTTTGGGG -u "???.....???" -b "" -k "?" -f 3 -e 1 reads.fastq
The UMIs will be added to the read ID in the output .fastq file and flexiplex_reads_barcodes.txt will contain a list of identified UMIs. Note that in the current version, the sequence provided after -u needs to be a string of wildcard characters, ‘?’ the length of the expected UMI. The example above comes from data we have tested on and may need to be adjusted for different library preparation kits.
Demultiplexing other read data by barcode
Flexiplex is highly flexible (as the name suggests) and the order, sequence and maximum distance to flanking, barcode and UMI sequence can all be manually set e.g. a barcode structure with UMI before barcode, and a constant sequence between them might look like:
flexiplex -x <left flank> -u "??????????" -x <constant sequence between UMI-barode> -b "????????????????" -x <right flank> -k <list of barcodes, or barcode_file> -f <flank maximum distance> -e <barcode maximum distance> reads.fastq > new_reads.fastq
Here -u and -b give a pattern of the expected UMI and barcode sequence, in this instance wildcards of length 10bp and 16bp respectively, the exact sequences of the barcodes are provided through -k. -e and -f which are the maximum barcode and flanking sequence edit distances respectively may also need to be adjusted. As a guide we use -e 2 for 16bp barcodes and -f 8 for 32bp (left+right) flanking sequence.
Demutiplexing multiple barcodes
Some barcoding schemes, such as visiumHD 3’, split-seq, pip-seq etc, embed multiple barcodes at either or both ends of a read. These are often (but not always) accompanied by short spacer sequences and a UMI. Barcoding structures like this are best handled with multiple runs of flexiplex - where each run searches for one the barcodes and adds it into the read header. Since flexiplex can read and write fastq format from standard IO, these multiple searches can achieved in a single command line using pipes e.g.:
flexiplex [options to find BC1 + UMI] | flexiplex [options to find BC2] | flexiplex [options to find BC3] > final_out.fastq
The read IDs in the final output will then have the following structure: @BC3#BC2#BC1_UMI#originalID
To improve the identification of the barcode region, all flexiplex runs prior to the last one should switch read trimming off (-r false)(version 1.02.6+ only). The order the barcodes are searched does not matter, aside from it impacting the order they are reported in the read header.
As a concrete example. Imgaine we have a barcode structure like this: [UMI][ACA][BC1][TCTCTC][BC2][GTGTGT][BC3][TTTTTTTTTT][cDNA], where the UMI and barcodes are all 8bp long. We could demultiplex with:
cat raw_reads.fastq |
flexiplex -u "????????" -x "ACA" -b "????????" -x "TCTCTC????????GTGTGT????????TTTTTTTTTT" -k barcodes.txt -r false |
flexiplex -x "ACA????????TCTCTC" -b "????????" -x "GTGTGT????????TTTTTTTTTT" -k barcodes.txt -r false |
flexiplex -x "ACA????????TCTCTC????????GTGTGT" -b "????????" -x "TTTTTTTTTT" -k barcodes.txt -r true > final_out.fastq
The edit distances for the flank sequence (-f) and barcode (-e) may need to be adjusted for the search length and number of barcodes. e.g. -e 1 would be reasonable when the barcodes are 8 bp long and there are ~1000 of them.
Simple search
Flexiplex can also be used to perform a simple error tolerant grep-like search of a single sequence, by define the sequence with -x and the required edit distance with -f. Matching reads will be printed to standard out.e.g.
flexiplex -x "CACTCTTGCCTACGCCACTAGC" -d grep -f 3 reads.fasta
Note, the the order of the -x and -d matter here. The search sequence needs to be defined before the -d preset is used.
Alternatively, the search sequence can be split so that a subsequence has a lower error tolerance e.g.:
flexiplex -r false false -x "CACTCTTGCC" -k "TACGC" -b "TACGC" -x "CACTAGC" -f 3 -e 0 reads.fasta
Here, we have defined that a perfect match (-e 0) is required for the middle (barcode) sequence, TACGC. Whereas the sequence surrounding this: CACTCTTGCC?????CACTAGC can have up to 3 mismatches/indels (-f 3). This is particularly useful for error tolerance searching of SNPs, as the alternative allele and can excluded through the requirement of a perfect match for the sequence at the variant.
Genotyping reads
An alternative on the simple search above, is to allow multiple alleles to be searched for e.g. to search for the KRAS variant c.34G>A run:
flexiplex -r false -x "GTATCGTCAAGGCACTCTTGCCTACGC" -b "CAC?AGC" -k "CACTAGC,CACCAGC" -x "TCCAACTACCACAAGTTTATATTCAGT" -e 0 -f 15 reads.fasta > kras_var_reads.fasta
Where -k here lists the mutant and wild type variants (reverse complimented), with a few bp either side, -x are the adjacent sequence left and right of these respectively and -b gives the pattern of the barcode, which may include wildcards (‘?’).
Genotyping cells - long reads
Searching/genotyping and demultiplexing can be chained together. e.g. assign cellular barcodes then search for a specific mutation:
flexiplex -d <chemistry> -k barcode_list.txt reads.fasta | flexiplex -n barcode_mutation_mapping -r false -x "GTATCGTCAAGGCACTCTTGCCTACGC" -k "CACTAGC,CACCAGC" -b "CAC?AGC" -x "TCCAACTACCACAAGTTTATATTCAGT" -e 0 -f 15 -u "" reads.fasta > kras_var_reads_with_barcodes.fasta
Assigning genotype to cells - short reads
Flexiplex can be also be used on 10x short read data to search for cells with a specific target sequence such as a mutation or fusion of interest from the raw read data. Here you “paste” the two read ends together and run in a similar way as you would for long reads. e.g. To search for a variant in 10x 3’ data:
paste Sample_R1.fastq Sample_R2.fastq | \
sed "/^[@+]/! s/\t/TTTTT/g" | \
sed "/^[@+]/! s/^/CTACACGACGCTCTTCCGATCT/g" | \
flexiplex -x <variant sequence (20-40bp)> -d grep | \
flexiplex -d 10x3v3
This prefixes the read (before the barcodes) with the 10x primer (CTACACGACGCTCTTCCGATCT) and adds some polyT sequence after the UMI (to recreate the long-read-like sequence structure). It then searches for the variant, then searches for the barcode immediately after the primer and returns a list of barcodes with the variant in flexiplex_barcodes_counts.txt. The read IDs can be found in flexiplex_reads_barcodes.txt
In this example we assume no sequencing errors in the barcodes as the data is Illumina, however a barcode list could also be provided (to the final command) to error correct. The order of demuliplexing and searching can also be switched. e.g.:
paste Sample_R1.fastq Sample_R2.fastq | sed "/[@+]/! s/^/CTACACGACGCTCTTCCGATCT/g" | flexiplex -d 10x3v3 -k barcodes_list.txt | flexiplex -x <variant sequence> -d grep
Output
New reads file
Read with a matching barcode will be reported to standard output (or to individual files if the -s true option is provided).
If read chopping and ID replacement is used (-r true, -l true default):
- Read IDs will be replaced with the following format (similar to FLAMES):
_ # _<+/-> of where <+/-> indicates whether the barcode was found on the forward or reverse strand of the original read, M is the number of barcodes found in direction indicated by +/- and N is the 1st, 2nd etc. of those barcode. - Reads will be reverse complimented if the barcode was found in the reverse direction. For 10x 3’ data this puts all reads in the reverse direction of the mRNA (3’->5’)
- If multiple barcodes are found in the same direction the read is split at the position of the second or subsequent primer, and multiple reads reported.
- The primer+barcode+umi+polyT sequence is removed from the read. If barcodes are found in both the forward and reverse directions on a read, the same read would be reported multiple time (once forward and once reverse). To overcome this duplication, data can be mapped as stranded
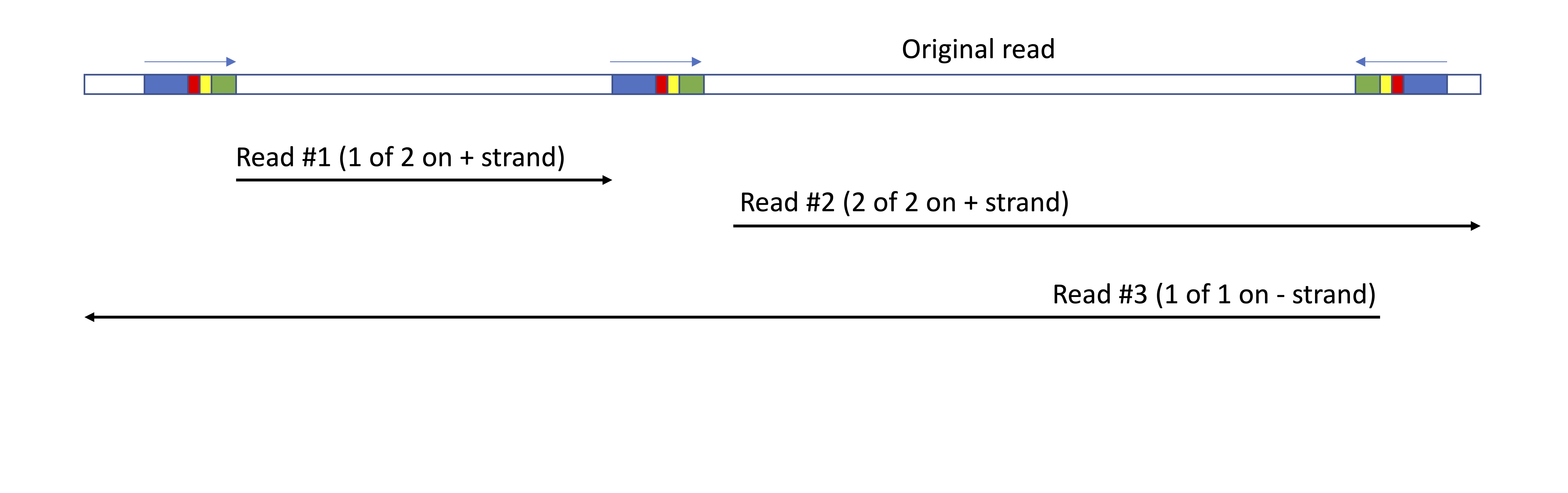 Schematic of default behaviour if multiple barcodes are identified in a read
Schematic of default behaviour if multiple barcodes are identified in a read
If read chopping and ID replacement is not used (-r false, -l false):
- Any read with matching flank and barcode sequence will be reported with read ID appended with _<+/-> as above.
- Read is reported only once even if multiple flank/barcode sequences found
- If a flank/barcode is only found in the reverse direction, the read will be reverse complimented.
Table of barcodes found for each read
This will be a file called flexiplex_reads_barcodes.txt or
Read CellBarcode FlankEditDist BarcodeEditDist UMI
SRR12282458.3 CTCAGAAGTTTGCATG 5 0 TTCTTAGCGA
SRR12282458.4 CGACTTCAGCTGTCTA 1 0 ACCGCATCCG
SRR12282458.6 GTAGGCCCAAGGCTCC 0 0 ATTATATCCT
SRR12282458.8 TCTCATATCACTCCTG 2 1 GAGCCCTGGG
SRR12282458.9 GTAGTCATCGTTTATC 1 0 TGCTGTATAG
SRR12282458.10 TGACTTTGTATAATGG 1 0 CCCTAATATT
...
Table of barcode frequency
This will be a file called flexiplex_barcodes_counts.txt or
It lists the number of reads that each barcode was found in, ordered by the more frequent. e.g. for a small dataset
GATCGATTCATCGCTC 15
AACCATGCACCTTGTC 12
ACGTCAAGTTTAAGCC 11
GTAACTGCATTGGGCC 11
AAGGCAGCATGGGAAC 10
ATGCGATTCACCTTAT 10
GATGCTAGTACAGCAG 10
GCAGCCATCATGCATG 10
GTTCATTCAATGTTGC 10
...
If the approximate number of cells is estimated, the top of this list can be used to generate a list of known barcodes e.g.
head -n <number of cell> flexiplex_barcodes_counts.txt > known_barcodes.txt
Then flexiplex run with -k known_barcodes.txt to more accurately assign the barcodes to reads (note that the second column in known_barcodes.txt gets ignored).
Table of the number of barcode at each barcode frequency
This is printed to standard output when no barcodes are provided (ie. flexiplex is in barcode discovery mode and -k not provided). e.g. for a small dataset:
Reads Barcodes
15 1
14 0
13 0
12 1
11 2
10 5
9 6
8 13
7 22
6 17
5 29
4 62
3 86
2 124
1 1867
The first line can be interpreted as there was 1 barcode which was found in 15 reads etc. This data is provided to help work out how many cells were sequenced (e.g. by creating a knee plot).
Tips to speed up flexiplex
Flexiplex can be slow if checking against a large number of barcodes (>1000) in noisy reads. Below are a few ideas you can try to speed up barcode demultiplexing:
- Run flexiplex in barcode discovery mode, then intersect the found barcodes with the known list to reduce the search space.
- Flexiplex now supports multi-threaded, but can still be I/O limited. You may be able to achieve faster parallel excecution by splitting the .fasta or .fastq into several smaller files and running flexiplex on each in parallel. The split linux command can help split files up. Passing flexiplex a read filename, rather than the reads through stdin, may also be faster on some platforms.
- Reduce the tolerated edit distance of the flanking and barcode sequence (-f and -e flags)
Support or Contact
To report issues, provide feedback or make a request please post a new github issue. We are keen to hear if you are using flexiplex for a use case not described above or if you have suggestions for predefined settings/options (e.g. bulk sample demultiplexing).